





-
試験デザイン
海外第Ⅲ相臨床試験(PALACE-1/2/3/4試験)1-11)海外データ)
活動性乾癬性関節炎患者を対象とした4つの海外第Ⅲ相臨床試験(PALACE-1試験、PALACE-2試験、PALACE-3試験及びPALACE-4試験)により、本剤20mg及び30mgを1日2回投与したときの有効性及び安全性を評価した。なお、これら4試験の試験デザインは同一であった。
PALACE各試験の違いはこちら
【目的】
オテズラの2用量(20又は30mg)を1日2回経口投与したときの活動性乾癬性関節炎に対する有効性及び安全性を、プラセボを対照として評価する。
【対象】
PALACE-1/2/3試験:乾癬性関節炎の確定診断から6ヵ月以上経過し、低分子又は生物学的疾患修飾性抗リウマチ薬(DMARDs)による治療にもかかわらず活動性乾癬性関節炎※3を有する患者1,493例(PALACE-1試験:504例、PALACE-2試験:484例、PALACE-3試験:505例)
PALACE-4試験:乾癬性関節炎の確定診断から3ヵ月以上経過し、低分子又は生物学的DMARDsによる治療歴がなく活動性乾癬性関節炎※3を有する患者527例
【方法】
第Ⅲ相多施設共同、並行群間試験。本試験はプラセボ対照期(24週間、ランダム化、二重盲検)、実薬投与期(最短28週間、ランダム化、二重盲検)及び長期安全性評価期(最長4年間、非盲検)の3期で構成された。
対象をオテズラ20mg 1日2回投与群、オテズラ30mg 1日2回投与群又はプラセボ群のいずれかに1:1:1でランダムに割付け、24週間投与した。16週時に圧痛関節数又は腫脹関節数が20%以上改善しなかった場合は早期離脱とし、盲検下で実薬を投与した。プラセボ群の早期離脱例は、オテズラ20mg又はオテズラ30mg 1日2回投与のいずれかに盲検下で1:1で再割付けした。オテズラ20mg群及び30mg群の早期離脱例は、同用量のオテズラ投与を盲検下で継続した。24週時にプラセボ群の患者をオテズラ20mg又は30mg 1日2回投与のいずれかに盲検下で1:1で再割付けし、オテズラ20mg 1日2回投与群及び30mg 1日2回投与群の患者は、盲検下で同用量の投与を継続した。
【主要評価項目】
投与16週時のACR20達成率※4(検証的な解析項目)
【副次評価項目】
投与16週時のACR50/70達成率、投与16週時のACRコンポーネントスコアのベースラインからの変化率、投与16、24週時のDSS※5のベースラインからの変化量、投与16、24週時のMASES※6のベースラインからの変化量、投与16、24、52 週時のDSS 0又はMASES 0達成率、投与16週、24週時のHAQ-DIスコア※7のベースラインからの変化量、投与52週時のDSS 0又はMASES 0達成率の推移等
【その他の評価項目】
投与16、24、52週時の圧痛関節数及び腫脹関節数のベースラインからの変化率、投与260週時までの圧痛関節数及び腫脹関節数のベースラインからの変化率、投与260週時までのDSS 0又はMASES 0達成率の推移
【解析計画】
目標症例数は主要評価項目(投与16週時のACR20達成率)においてプラセボ群に対する差を検出できるよう設計した。主要評価項目並びに投与16週時及び24週時の副次評価項目は、オテズラの各用量とプラセボとの間で比較検定を実施した。有意水準0.05に制御するため、最初に主要評価項目について統計学的検定を実施し、その後、投与16週時及び24週時の副次評価項目を階層的に実施した。また、16週及び24週のACR変化率の各部分集団の解析にはANCOVAによるノンパラメトリック法を用いた。活動性PsA治療でのオテズラの臨床的有効性を示すために、事前にPALACE-1試験、PALACE-2試験及びPALACE-3試験の併合解析を計画し、指趾炎(ベースラインのDSS>0)及び/又は付着部炎(ベースラインのMASES>0)を有する患者に対するロバストな解析を実施した。安全性の併合解析については、PALACE-1試験、PALACE-2試験、PALACE-3試験、PALACE-4試験を統合した。
- オテズラの投与開始時は、最初の6日間にオテズラを1日あたり10mgずつ漸増投与。
- 16週時に圧痛関節数又は腫脹関節数が20%以上改善しなかった場合は早期離脱とし、盲検下で実薬を投与。
- 圧痛関節数3以上及び腫脹関節数3以上。
- ACR20達成率:ACR(American College of Rheumatology)のコアセットのうち、①圧痛関節数(78関節)及び②腫脹関節数(76関節)が20%以上改善、かつ③患者による疾患活動性の全般評価[PGA]、④医師による疾患活動性の全般評価[PhyGA]、⑤患者による疼痛評価、⑥Health Assessment Questionnaire-Disability Index(HAQ-DI)及び⑦C-反応性タンパク(CRP)の5項目のうち3項目で20%以上改善した患者割合。
ACR50/ACR70達成率:同様に50%/70%以上改善した患者割合。 - DSS:指趾炎が認められた手足の指の数を0~20点で評価する。
- MASES:13の腱付着部のうち痛みがある部位の数を0~13点で評価する。
- HAQ-DI (Health Assessment Questionnaire-Disability Index):20の質問からなる自己評価式の尺度。患者の機能的能力の困難度を4段階で評価し、得点が高いほど困難度が高いことを示す。
注) 本項ではPALACE-4試験については24週でカットオフしたデータに基づく治験総括報告書から試験成績を紹介する(承認時)。PALACE-1/2/3試験についてはKavanaugh A et al.:Arthritis Res Ther. 21:118, 2019から、最大260週まで投与したデータを紹介する。
また、オテズラ20mg 1日2回投与は本邦未承認の用法・用量のため、有効性についてはオテズラ30mg 1日2回投与時の結果のみ記載する。4. 効能又は効果(一部抜粋)
○局所療法で効果不十分な尋常性乾癬 ○乾癬性関節炎
6. 用法及び用量
通常、成人にはアプレミラストとして以下のとおり経口投与し、6日目以降はアプレミラストとして1回30mgを1日2回、朝夕に経口投与する。
1日目 2日目 3日目 4日目 5日目 6日目以降 朝 朝 夕 朝 夕 朝 夕 朝 夕 朝 夕 10mg 10mg 10mg 10mg 20mg 20mg 20mg 20mg 30mg 30mg 30mg -
患者背景
PALACE-1/2/3試験(併合解析) PALACE-4試験 プラセボ群
(n=496)オテズラ30mg
1日2回投与群
(n=497)プラセボ群
(n=176)オテズラ30mg
1日2回投与群
(n=176)年齢(歳)、平均値±SD 50.6±11.6 50.6±11.4 50.5±11.6 48.4±12.5 女性、n(%) 256 (51.6) 275 (55.3) 86 (48.9) 96 (54.5) 体重(kg)、平均値±SD 86.4±21.1 84.5±19.6 82.4±18.2 85.7±20.6 BMI(kg/㎡)、平均値±SD 30.0±6.5 29.7±6.2 28.7±5.6 29.7±6.4 乾癬性関節炎の罹病期間(年)、
平均値±SD7.3±7.3 7.5±7.8 3.4±5.1 3.6±5.0 腫脹関節数(0-76)、平均値±SD 11.0±8.0 11.6±8.2 11.3±7.6 10.9±8.6 圧痛関節数(0-78)、平均値±SD 19.9±14.8 21.9±15.2 19.6±13.7 19.5±14.4 DSS※1 n 205 221 90 84 平均値±SD 3.2±3.3 3.3±3.3 3.1±2.5 3.5±3.3 MASES※2 n 331 327 115 111 平均値±SD 4.8±3.3 4.5±3.2 3.6±2.6 3.7±3.0 ※1 ベースラインで指趾炎を有していた患者のデータ
※2 ベースラインで付着部炎を有していた患者のデータ
-
PALACE-4試験 有効性
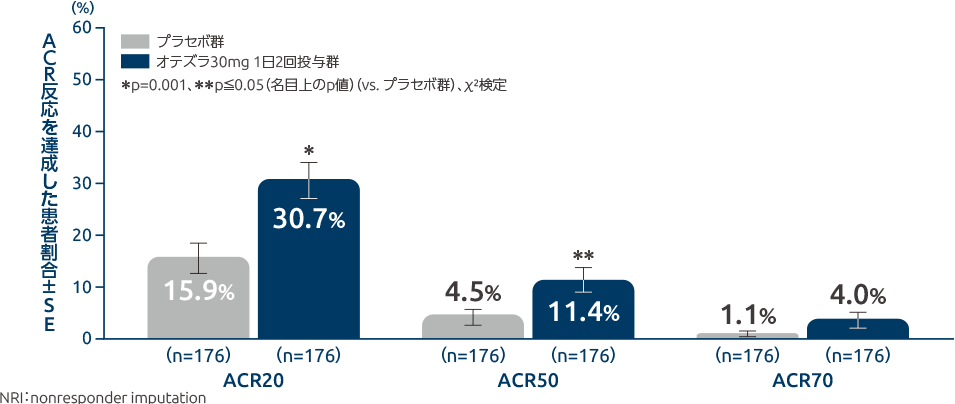
投与16週時のACR20達成率はプラセボ群に比べオテズラ群で高く、疾患活動性が改善しました
投与16週時のACR20/50/70達成率
[ACR20達成率:主要評価項目(検証的な解析結果)、ACR50/70達成率:副次評価項目]オテズラ30mg 1日2回投与群の投与16週時のACR20、ACR50、ACR70達成率はそれぞれ30.7%、11.4%、4.0%であり、ACR20およびACR50達成率はプラセボ群と比べて統計学的に有意に高い結果でした[p=0.001、p≦0.05(名目上のp値)、χ2検定]。
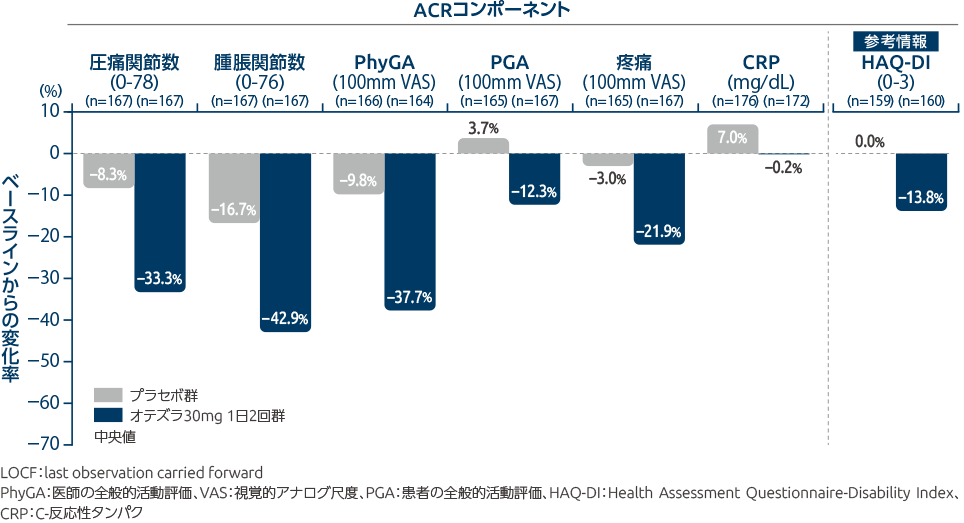
投与16週時のACRコンポーネントスコアのベースラインからの変化率(副次評価項目)
オテズラ30mg 1日2回投与群では、投与16週時の各ACRコンポーネントスコアの変化率は以下の通りでした。
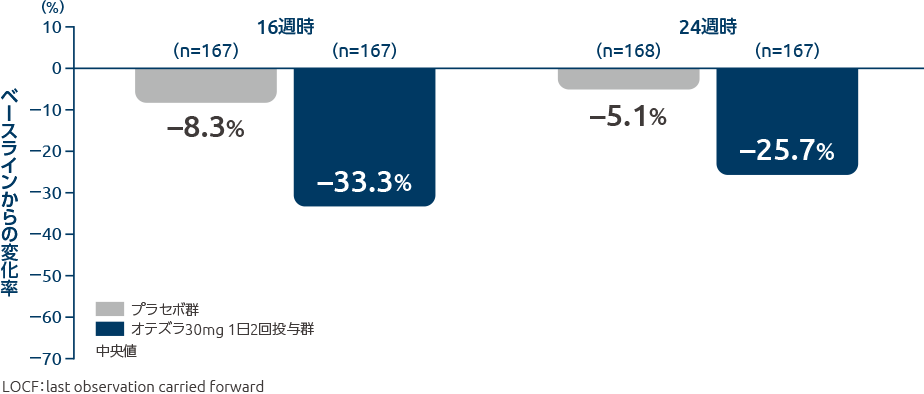
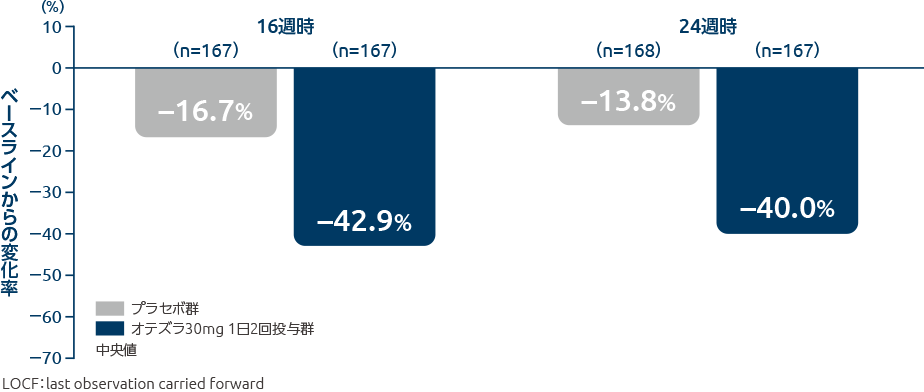
オテズラ錠の投与により、圧痛関節数及び腫脹関節数のベースラインからの変化率は-20%より低い値でした
投与16週時、24週時の圧痛関節数のベースラインからの変化率(その他の評価項目)
オテズラ30mg 1日2回投与群の圧痛関節数のベースラインからの変化率(中央値)は、投与16週時で-33.3%、投与24週時で-25.7%でした。
投与16週時、24週時の腫脹関節数のベースラインからの変化率(その他の評価項目)
オテズラ30mg 1日2回投与群の腫脹関節数のベースラインからの変化率(中央値)は、投与16週時で-42.9%、投与24週時で-40.0%でした。
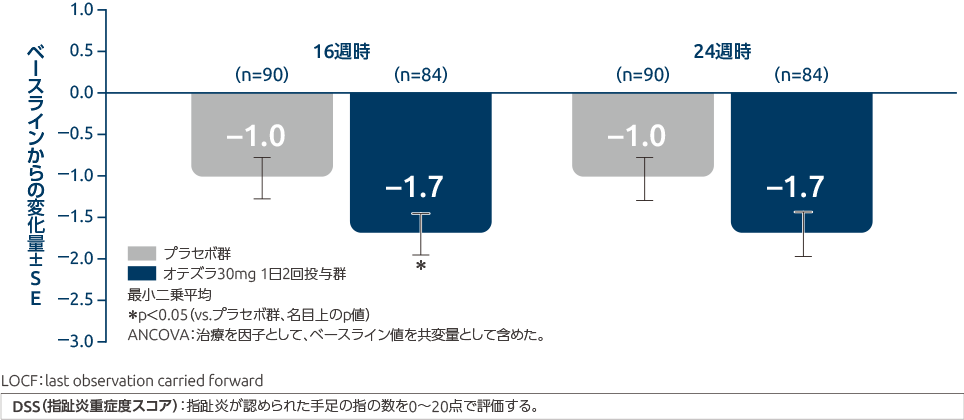
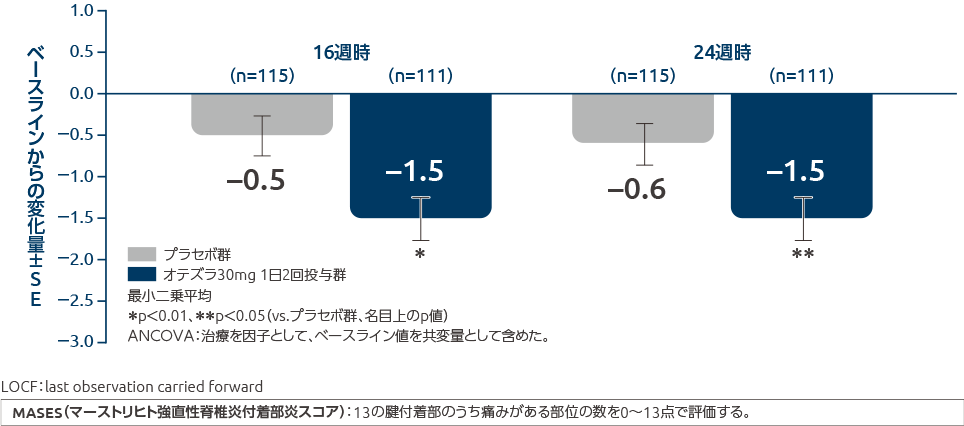
オテズラ錠の投与により、指趾炎重症度スコア及び付着部炎スコアはプラセボ群と比べて低くなりました
投与16週時、24週時のDSSのベースラインからの変化量(副次評価項目)
ベースラインで指趾炎を有していた患者において、オテズラ30mg 1日2回投与群のDSSのベースラインからの変化量(最小二乗平均)は、投与16週時で-1.7と指趾炎の症状が改善し(p<0.05、名目上のp値、ANCOVA)、投与24週でも-1.7でした。
投与16週時、24週時のMASESのベースラインからの変化量(副次評価項目)
ベースラインで付着部炎を有していた患者において、オテズラ30mg 1日2回投与群のMASESのベースラインからの変化量(最小二乗平均)は、投与16週時で-1.5、投与24週時で-1.5と付着部炎の症状が改善しました( p<0.01、p<0.05、名目上のp値、ANCOVA)。
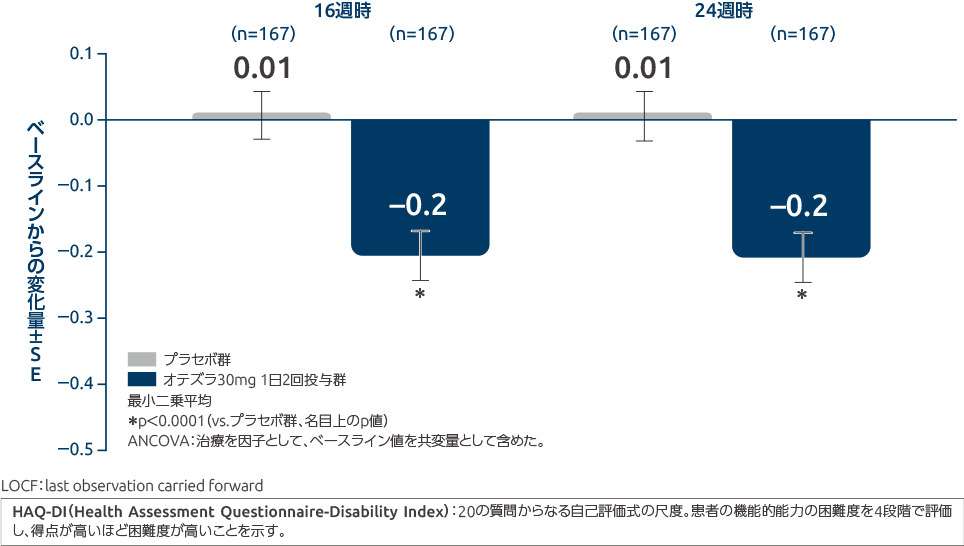
参考情報 投与16週時、24週時のHAQ-DIスコアのベースラインからの変化量(副次評価項目)
オテズラ30mg 1日2回投与群のHAQ-DIスコアのベースラインからの変化量(最小二乗平均)は、投与16週時で-0.2、投与24週時で-0.2でした。
4. 効能又は効果(一部抜粋)
○乾癬性関節炎 -
PALACE-1/2/3試験 有効性
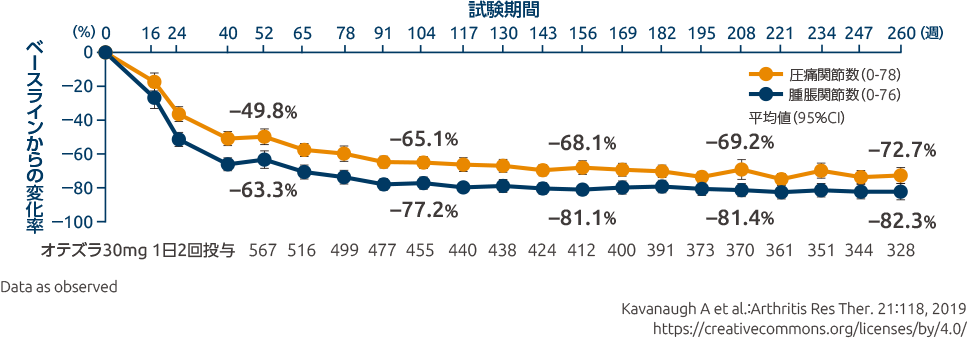
オテズラ錠の投与により、乾癬性関節炎の疾患活動性の指標は以下の通りに推移しました
投与260週時までの圧痛関節数及び腫脹関節数のベースラインからの変化率
(52週時:その他の評価項目、260週時まで:その他の評価項目)オテズラ30mg 1日2回投与群における圧痛関節数のベースラインからの変化率(平均値)は、投与52週時では-49.8%であり、投与260週時では-72.7%でした。
同様に、腫脹関節数のベースラインからの変化率(平均値)は、投与52週時では-63.3%であり、投与260週時では-82.3%でした。
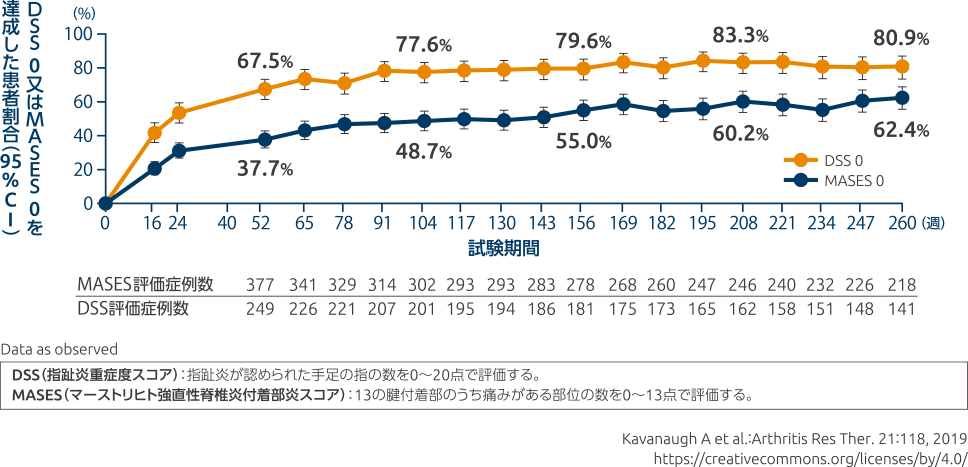
投与260週時までのDSS 0又はMASES 0達成率の推移
(52週時:副次評価項目、260週時まで:その他の評価項目)
ベースラインで指趾炎を有していた患者におけるDSS 0達成率は、投与52週時では67.5%であり、投与260週時では80.9%でした。
同様に、ベースラインで付着部炎を有していた患者におけるMASES 0達成率は、投与52週時では37.7%であり、投与260週時では62.4%でした。
4. 効能又は効果(一部抜粋)
○乾癬性関節炎 -
安全性
投与24週時までの副作用発現率はオテズラ30mg 1日2回投与群33.1%、プラセボ群14.2%で、主な有害事象は悪心、下痢、頭痛でした
プラセボ対照期(0~24週)の安全性の概要
PALACE-4試験における副作用又は有害事象発現状況は以下の通りでした。
プラセボ対照期(0週時~24週時)
n(%) プラセボ群※1
(n=176)オテズラ30mg
1日2回投与群※2
(n=175)副作用 25 (14.2) 58 (33.1) 主な有害事象(いずれかの群で5%以上発現) 悪心 4 (2.3) 28 (16.0) 下痢 3 (1.7) 21 (12.0) 頭痛 4 (2.3) 15 (8.6) 重篤な有害事象※3 5 (2.8) 1 (0.6) 投与中止に至った有害事象※4 4 (2.3) 6 (3.4) - 16週時に早期離脱しなかった患者の0~24週までのデータ、及び16週目に早期離脱した患者の0~16週時までのデータが含まれる
- 早期離脱に関係なく、オテズラ30mg 1日2回投与群に無作為割付けされたすべての患者の0~24週時までのデータが含まれる
- 重篤な有害事象
・プラセボ群:糖尿病性壊疽、喀血、乾癬性関節症、子宮内膜増殖症、瘢痕ヘルニア各1例(0.6%)
・オテズラ30mg 1日2回投与群:急性腎盂腎炎1例(0.6%) - 2例以上に認められた投与中止に至った有害事象
・オテズラ30mg 1日2回投与群:頭痛3例(1.7%)、下痢、悪心各2例(1.1%)
オテズラ30mg 1日2回投与264週時までの有害事象発現率は85.6%で、主な有害事象は下痢、悪心などでした
オテズラ投与期(0~260週)及び観察追跡期(261~264週)の安全性の概要
PALACE-1/2/3/4試験(併合解析)における有害事象発現状況は以下の通りでした。
オテズラ投与期(0~260週)n(%) オテズラ30mg 1日2回投与群※5 (n=973) 有害事象 833 (85.6) 主な有害事象(5%以上) 下痢 166 (17.1) 悪心 164 (16.9) 上気道感染 140 (14.4) 頭痛 129 (13.3) 鼻咽頭炎 120 (12.3) 高血圧 86 (8.8) 気管支炎 80 (8.2) 尿路感染症 69 (7.1) 副鼻腔炎 65 (6.7) 咳嗽 56 (5.8) 背部痛 53 (5.4) 重篤な有害事象※6 180 (18.5) 投与中止に至った有害事象※7 116 (11.9) 死亡に至った有害事象※8 4 (0.4) - 0週にオテズラ30mg 1日2回投与群であった患者及び16週又は24週にプラセボ群からオテズラ30mg 1日2回投与に切り替えた患者
- 重篤な有害事象
乾癬性関節症13例(1.3%)、変形性関節症6例(0. 6%)、胆石症、鼡径ヘルニア、冠動脈疾患各5例(0. 5%)、狭心症、心房細動各4例(0. 4%)、肺炎、不安定狭心症、乳癌各3例(0.3%)、乾癬、発熱各2例(0.2%)、うつ病、急性心筋梗塞、一過性脳虚血発作各1例(0.1%) - 投与中止に至った有害事象
下痢22例(2. 3%)、悪心20例(2. 1%)、頭痛15例(1.5%)、嘔吐8例(0. 8%)、上腹部痛、疲労、浮動性めまい各4例(0. 4%)、不眠症3例(0. 3%)、うつ病、不安各2例(0. 2%) - 死亡に至った有害事象(各1例)
・ オートバイ事故に関連した胸部及び頭部の外傷
・ 治験薬との関連はないと考えられる前腹壁の壊死性筋膜炎、難治性低血圧ショック、急性腎不全。患者には糖尿病の既往歴がありました
・ 治験薬との関連はないと考えられる脳血管障害。患者には脳血管障害、冠動脈疾患、不整脈、心筋梗塞、高血圧、脂質異常症、アテローム性動脈硬化症、冠動脈ステント留置、心房細動の既往歴がありました
・ 治験薬との関連はないと考えられる脳梗塞。患者には骨粗鬆症、閉経後、慢性アルコール中毒、貧血、高血圧の既往歴がありました
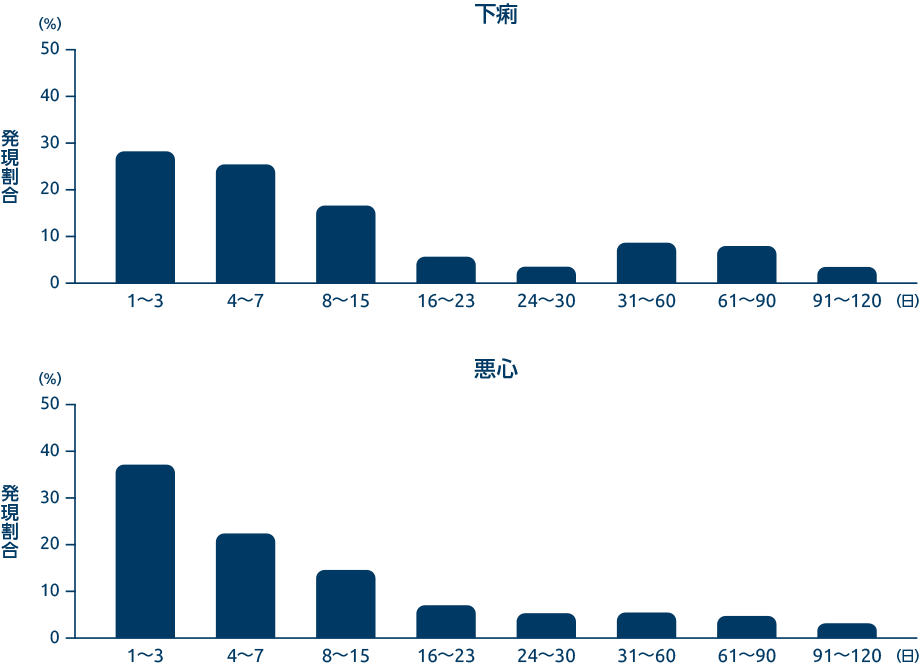
PALACE-1/2/3/4試験(併合解析)における下痢及び悪心の発現時期(有害事象)は以下の通りでした
乾癬性関節炎を対象とした海外第Ⅲ相臨床試験(PALACE-1/2/3/4試験併合)での下痢及び悪心の発現時期※
下痢、悪心、嘔吐、腹痛などの消化管障害は、PDE4阻害剤使用時にみられる事象であり、本剤の投与中にも発現することが報告されています。
また、市販後に重度の下痢の副作用が報告されています。
国内外の臨床試験において報告されたほとんどの下痢及び悪心の有害事象は投与開始後2週間以内に発現し、4週間以内に消失しました。
- 本剤の投与開始時は「用法及び用量」を遵守し、漸増投与を行うように指導してください。
- 消化管障害が軽微な場合には、経過観察してください。
- 症状が重篤な場合や症状の改善が認められない場合には、適宜対症療法を実施するとともに、本剤を中止する等の適切な処置を行ってください。
※オテズラ20mg 1日2回投与は本邦未承認用法及び用量のため、承認された用法及び用量であるオテズラ30mg 1日2回投与での発現割合を示します。
4. 効能又は効果(一部抜粋)
○乾癬性関節炎

-
References
- CC-10004-PSA-002試験(承認年月:2016年12月、CTD2.7.6.25)(承認時評価資料)
- Kavanaugh A et al.:Ann Rheum Dis. 73:1020-1026, 2014
- Kavanaugh A et al.:J Rheumatol. 42:479-488, 2015
- CC-10004-PSA-003試験(承認年月:2016年12月、CTD2.7.6.26)(承認時評価資料)
- CC-10004-PSA-004試験(承認年月:2016年12月、CTD2.7.6.27)(承認時評価資料)
- Edwards CJ et al.:Ann Rheum Dis. 75:1065-1073, 2016
- Amgen社社内資料(承認時評価資料):アプレミラストの海外第Ⅲ相臨床試験3試験の有効性併合解析
- Kavanaugh A et al.:Arthritis Res Ther. 21:118, 2019
- CC-10004-PSA-005試験(承認年月:2016年12月、CTD2.7.6.28)(承認時評価資料)
- Amgen社社内資料(承認時評価資料):アプレミラストの海外第Ⅲ相臨床試験4試験の安全性併合解析
- Wells AF et al.:Rheumatology(Oxford). 57:1253-1263, 2018
- Amgen社社内資料:アプレミラストの海外第Ⅲ相臨床試験の長期安全性併合解析
- Amgen社社内資料:アプレミラストの海外第Ⅲ相臨床試験3試験の安全性併合解析
利益相反:PALACE-1試験、PALACE-2試験、PALACE-3試験及びPALACE-4試験はAmgen社(旧 Celgene社)の資金提供によりアプレミラストの開発治験として実施され、この結果を報告した論文、2)[Kavanaugh A et al.: Ann Rheum Dis. 73:1020-1026, 2014]、3)[Kavanaugh A et al.: J Rheumatol. 42:479-488, 2015]、6)[Edwards CJ et al.: Ann Rheum Dis. 75:1065-1073, 2016]、8)[Kavanaugh A et al.: Arthritis Res Ther. 21:118, 2019]、11)[Wells AF et al.: Rheumatology(Oxford). 57:1253-1263, 2018]の著者らには、Amgen社(旧 Celgene社)の社員や指導料などの謝金を受領した者を含みます。

