



試験概要1-4)
国際共同第Ⅱ相試験(DeLLphi-301試験)
目的
2つ以上の化学療法(うち、少なくとも1つは白金系抗悪性腫瘍剤を含む)歴のある小細胞肺癌患者を対象に、イムデトラの有効性、安全性、忍容性及び薬物動態を評価する
対象
2つ以上の化学療法(うち、少なくとも1つは白金系抗悪性腫瘍剤を含む)歴のある小細胞肺癌患者220例
試験デザイン
国際共同第Ⅱ相、無作為化用量評価、非無作為化用量拡大、非盲検、多施設
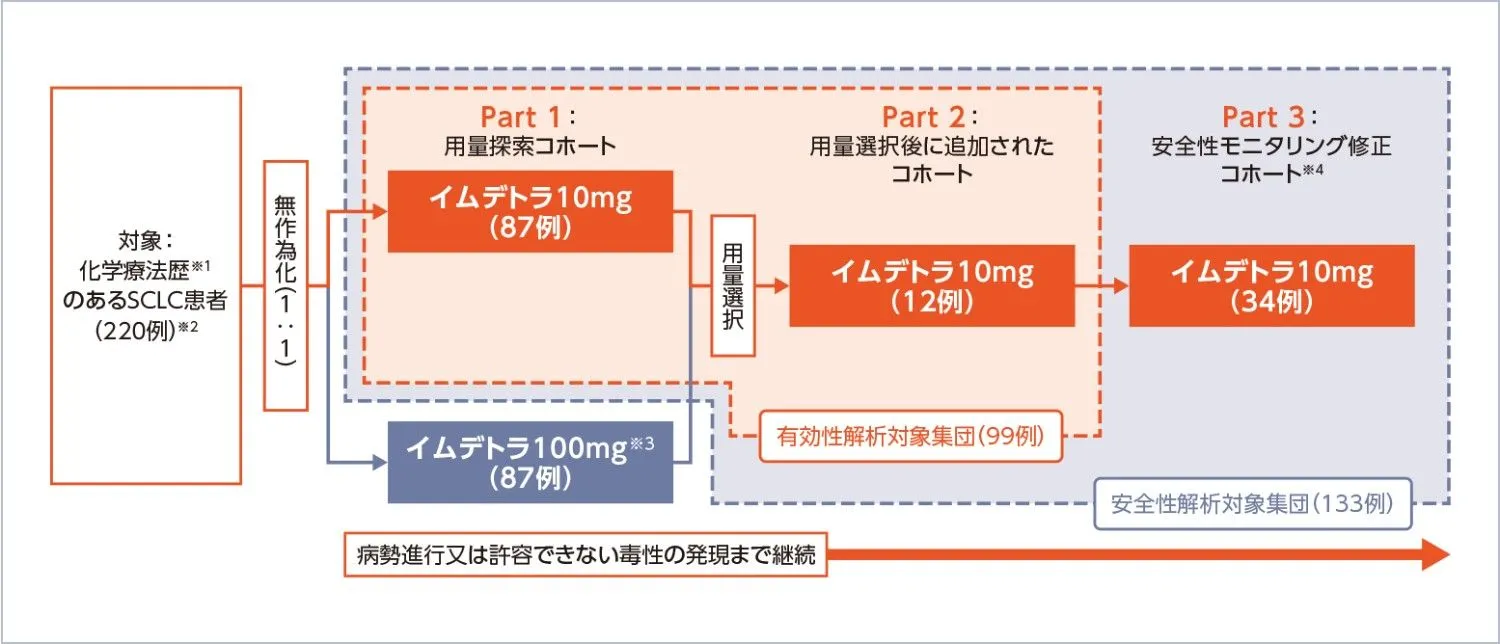
投与方法:
イムデトラ1mgをサイクル1の1日目に、10mg又は100mgをサイクル1の8日目及び15日目、その後2週間間隔で投与する。デキサメタゾン8mg(又はその他のコルチコステロイドの等価用量)はサイクル1の1日目及び8日目のみ、イムデトラの投与前1時間以内に静脈内投与する。
※1:
2つ以上の化学療法(うち、少なくとも1つは白金系抗悪性腫瘍剤を含む)
※2:
試験に組み入れられた222例のうちイムデトラが投与されなかった2例を除く220例が評価対象とされた。
※3:
本邦における承認外の用法及び用量のため、100mg群の解析結果は省略する。
※4:
サイクル1の1日目及び8日目において、Part1及び2ではイムデトラ投与後48時間の入院が必要とされたが、Part3では24時間に短縮された。
主要評価項目
【Part 1及び2】客観的奏効率(ORR)[BICRによるmodified RECIST ver.1.1(mRECIST ver.1.1)に基づく]
【Part 3】安全性
副次評価項目
【全Part】奏効期間(DOR)[BICR]、病勢コントロール率(DCR)[BICR]、病勢コントロール期間(DC期間)
[BICR]、無増悪生存期間(PFS)[BICR]、全生存期間(OS)、安全性、忍容性、薬物動態、免疫原性など
主な選択基準
- 18歳(又は法定成人年齢)以上で、組織学的又は細胞学的に確定診断された再発又は難治性SCLCの男女
- 1つのプラチナ製剤ベースレジメン及び1ライン以上の他の治療後に進行又は再発した患者※
- ECOG PS 0~1
- イムデトラ初回投与前21日以内にmRECIST ver.1.1で規定される測定可能病変を有し、十分な臓器機能を有する患者
- イムデトラ初回投与の少なくとも2週間前に脳転移に対する根治的治療が完了しており、病変の進行を認めない
※:
[1]
プラチナ製剤ベースレジメンによる再治療は二次治療とみなした。
[2]
プラチナ製剤ベースレジメンと、それに続く維持療法としての免疫チェックポイント阻害剤/抗PD-L1抗体の投与は1ラインとみなした。
[3]
標準治療の一次全身治療にプラチナ製剤を含む化学療法とPD-L1阻害剤の併用療法が含まれる国では、一次全身治療でのPD-L1阻害剤が無効又は不適応であったこととした。
主な除外基準
- 未治療又は症候性の脳転移及び軟膜疾患、間質性肺疾患又は活動性の非感染性肺炎の所見がある
- 再発性肺炎(Grade 2以上)、重篤で生命を脅かす免疫介在性有害事象又は投与関連反応(免疫腫瘍薬による治療中に恒久的な中止に至ったものを含む)を発現した患者
- 前治療による未回復毒性(CTCAE ver.5.0におけるGrade 1又は適格基準で指示されたレベルに回復しない)がある
- 過去2年以内の他の悪性腫瘍の既往がある(根治治療が行われ過去2年以上再発を認めないもの、登録時点で疾患を認めないものを除く)
- イムデトラ初回投与前12ヵ月以内の心筋梗塞及び/又は症候性うっ血性心不全[ニューヨーク心臓協会(NYHA)クラスII超]又は動脈血栓症の既往がある
解析計画
- Part 1では、患者は10mg群又は100mg群のいずれかに1:1で無作為化され、Part 1の中間解析に基づきPart 2の用量が選択された。Part 2にはPart 1と合わせて約100例が登録されるまで患者を組み入れた。Part 3はPart 1及び2の組み入れ完了後に開始し、選択された用量に最大約30例の患者を追加で組み入れ、修正したサイクル1の安全性モニタリング基準を用いた。
- 臨床的に意味のあるORRの閾値を15%と設定し、Part 1及びPart 2で目標用量10mgを投与された患者100例でのORRが24%を超えたときに、その97.5%CIの下限値が15%を上回るようデザインされた。
- 主要解析は、全ての患者が組み入れられ、ベースライン後の初回の腫瘍評価から24週間以上の追跡調査を受ける機会を得た時点で実施し、BICRによるmRECIST ver.1.1に基づく治療効果判定に準拠した。
-
腫瘍縮小効果に関連した有効性評価項目の主解析はBICR Full Analysis Set(BICR FAS)を対象として行った。BICR FASは、Part 1又はPart 2におけるイムデトラを1回以上投与した全ての患者のうち、ベースラインにBICRによるmRECIST ver.1.1に基づく測定可能病変を1つ以上有する患者とした。OSの解析は安全性解析対象集団で実施した。
【主要評価項目】
ORRについては、最良総合効果(BOR)として完全奏効(CR)、部分奏効(PR)、安定(SD)、進行(PD)、評価不能(NE)のうちCR及びPRを達成した患者数と割合をClopper-Pearsonの正確な97.5%CIとともに要約した。ベースライン後の腫瘍評価を実施しなかった患者は非奏効例とみなした。
【副次評価項目】
DOR、PFS、OS及びDC期間については、中央値及び選択した四分位数に関してKaplan-Meier推定値及びBrookmeyer-Crowley法(1982)による95%CIで要約した。選択した評価時点(例:6ヵ月、9ヵ月、12ヵ月時点)の無イベント率を示した。95%CIはKalbfleisch and Prentice(1980)の方法を用いて推定した。DORは、BORとしてCR又はPRを達成した患者のみ算出した。DCRは、BORとしてCR、PR又はSDを達成した患者数と割合をClopper-Pearsonの正確な95%CIとともに要約した。
【事前に規定されたサブグループ解析】
年齢、人種、地域、前治療ライン数、PD-1又はPD-L1阻害剤による前治療の有無、ベースラインの標的腫瘍病変の径和、プラチナ製剤に対する感受性、ベースラインの脳転移又は肝転移の有無、DLL3のカットポイントについて、ORRのサブグループ解析を行った。また、日本人サブグループについて、ORR、DOR、DCR、DC期間、PFS、OS及び安全性の解析を行った。
【安全性】
安全性解析対象集団にはイムデトラを1回以上投与された全ての患者を含めた。有害事象として、イムデトラの初回投与以降、イムデトラの最終投与後47日又は試験終了日のいずれか早い方までに認められたものを評価した。注目すべき有害事象としてサイトカイン放出症候群(CRS)、神経学的事象[免疫エフェクター細胞関連神経毒性症候群(ICANS)を含む]の評価を行った。CRS及びICANSは米国移植細胞治療学会(ASTCT)の判定基準に基づき重症度判定し、それ以外の有害事象はCTCAE ver.5.0に基づき重症度判定した。
【追加解析】
その他の有害事象として、血球減少、間質性肺疾患の評価を行った。本評価は、事前に計画した解析ではないものの、承認申請資料として提出し、その承認審査過程で評価を受けた。
4.効能又は効果
がん化学療法後に増悪した小細胞肺癌
5.効能又は効果に関連する注意
本剤の一次治療における有効性及び安全性は確立していない。
6. 用法及び用量
通常、成人にはタルラタマブ(遺伝子組換え)として、1日目に1mg、8日目に10mgを1回、1時間かけて点滴静注する。15日目以降は1回10mgを1時間かけて2週間間隔で点滴静注する。
7.用法及び用量に関連する注意(一部抜粋)
7.2 本剤投与によるサイトカイン放出症候群を軽減するため、1日目及び8日目の本剤投与前1時間以内に副腎皮質ホルモン剤を静脈内投与すること。また、1日目、8日目及び15日目の本剤投与後に輸液を行うこと。[1.2.2、7.4、8.2、11.1.1参照]
患者背景1,3)
ベースラインの人口統計学的特性及び疾患特性
| table text | table text | 全体※1: 10mg群、例数(%) |
日本人※2: 10mg群、例数(%) |
|
|---|---|---|---|---|
| Part 1/Part 2 (n=99) |
Part 3 (n=34) |
Part 1/Part 2 (n=11) |
||
| 性別 | 男性 | 71 (71.7) | 24 (70.6) | 11 (100.0) |
| 女性 | 28 (28.3) | 10 (29.4) | 0 | |
| 人種 | アジア人 | 41 (41.4) | 2 (5.9) | 11 (100.0) |
| 黒人/アフリカ系アメリカ人 | 0 | 1 (2.9) | 0 | |
| 白人 | 57 (57.6) | 31 (91.2) | 0 | |
| その他 | 1 (1.0) | 0 | 0 | |
| 年齢 | 中央値(範囲) | 64.0歳 (35~82) | 65.5歳 (49~80) | 63.0歳 (43~77) |
| 18-64歳 | 51 (51.5) | 14 (41.2) | 7 (63.6) | |
| 65-74歳 | 38 (38.4) | 14 (41.2) | 3 (27.3) | |
| 75-84歳 | 10 (10.1) | 6 (17.6) | 1 (9.1) | |
| 85歳以上 | 0 | 0 | 0 | |
| ECOG PS | 0 | 26 (26.3) | 10 (29.4) | 4 (36.4) |
| 1 | 73 (73.7) | 24 (70.6) | 7 (63.6) | |
| 前治療ライン数 | 1 | 2 (2.0) | 0 | 0 |
| 2 | 65 (65.7) | 22 (64.7) | 7 (63.6) | |
| 3 | 18 (18.2) | 6 (17.6) | 1 (9.1) | |
| 3超 | 14 (14.1) | 6 (17.6) | 3 (27.3) | |
| 前治療ライン数中央値(範囲) | 2.0 (1~6) | 2.0 (2~6) | 2.0 (2~5) | |
| PD-1/PD-L1阻害剤による前治療あり | 73 (73.7) | 28 (82.4) | 4 (36.4) | |
| 現存する腫瘍に対する放射線治療歴あり | 77 (77.8) | 24 (70.6) | 11 (100.0) | |
| 病期 | 0期又はⅠ期 | 0 | 0 | 0 |
| Ⅱ期 | 1 (1.0) | 2 (5.9) | 0 | |
| Ⅲ期 | 5 (5.1) | 1 (2.9) | 1 (9.1) | |
| Ⅳ期 | 87 (87.9) | 28 (82.4) | 10 (90.9) | |
| 不明又は欠測 | 6 (6.1) | 3 (8.8) | 0 | |
| 転移 | あり | 97 (98.0) | 32 (94.1) | 11 (100.0) |
| なし | 2 (2.0) | 2 (5.9) | 0 | |
| 脳転移 | あり | 22 (22.2) | 4 (11.8) | 4 (36.4) |
| なし | 77 (77.8) | 30 (88.2) | 7 (63.6) | |
| 肝転移 | あり | 38 (38.4) | 12 (35.3) | 2 (18.2) |
| なし | 61 (61.6) | 22 (64.7) | 9 (81.8) | |
| プラチナ感受性 | 90日未満 | 27 (27.3) | 7 (20.6) | 2 (18.2) |
| 90日以上180日未満 | 22 (22.2) | 7 (20.6) | 2 (18.2) | |
| 180日以上 | 20 (20.2) | 9 (26.5) | 2 (18.2) | |
| 不明 | 30 (30.3) | 11 (32.4) | 5 (45.5) | |
※1:
有効性解析対象集団(99例)の10mg群(Part 1及びPart 2)では16例(16.2%)、安全性解析対象集団(133例)の10mg群では23例(17.3%)が限局型SCLCに対する化学療法後に疾患進行した患者であった。
※2:
Part 3に組み入れられた日本人はいなかった。
-
References
- 社内資料:国際共同第Ⅱ相試験(DeLLphi-301試験/20200491試験)(承認時評価資料:CTD2.7.6.2)
- Ahn MJ et al. N Engl J Med. 2023; 389: 2063-2075.
[利益相反:本試験はAmgen社の支援により行われた。] - 社内資料:日本人部分集団における有効性(承認時評価資料:CTD2.7.3.3.4)
- 社内資料:日本人部分集団における安全性(承認時評価資料:CTD2.7.4.5.3)




IMD254116RM2|2026年6月作成